Développement des biosimilaires
Comment "copier" les biomédicaments ?
En premier lieu, des tests vont être réalisés sur le médicament biologique de référence.
Les CQA (critical quality attribute) vont être identifiés : ce sont des caractéristiques physiques, chimiques, biologiques... qui vont devoir être conservés. Des seuils de variation acceptables de ces propriétés vont être définis, sur la base de plusieurs lots du produit de référence.
Les lignées cellulaires vont être établies, avec l'insertion du gêne codant la protéine d’intérêt.
Ces protéines vont être comparées au produit de référence. Celles qui correspondent vont être identifiées, et les lignées cellulaires qui en sont à l'origine vont être retenues. Des banques de cellules vont être établies, pour production.
La production va changer d'échelle. Il va être vérifié que l'uniformité structurale et fonctionnelle des cellules est maintenue.
De nombreux lots du biomédicament de référence et du candidat biosimilaire vont être caractérisés pour vérifier la similarité.
(Source : "Developing Biosimilars" - AMGEN)
Fondamental : Données nécessaires
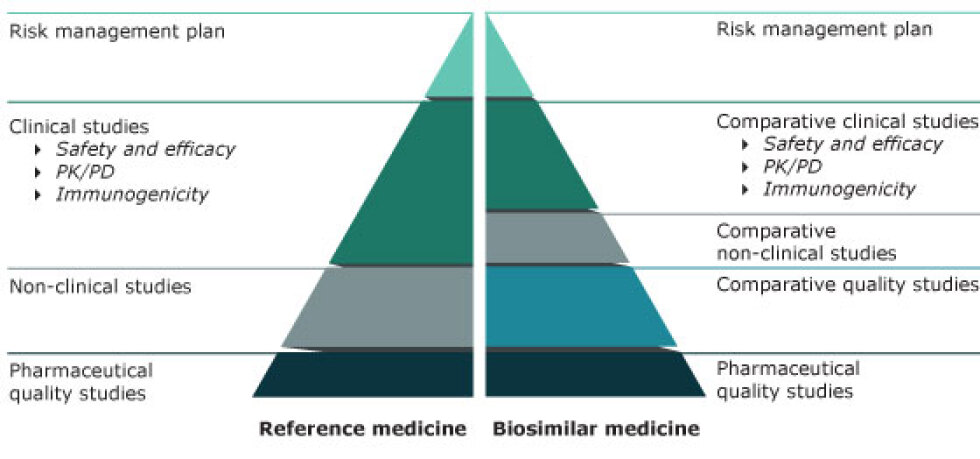
Pour être mis sur le marché, un médicament biosimilaire va devoir présenter une qualité, une tolérance et une sécurité similaires au biomédicament de référence.
Ainsi, la structure, la fonction, l'activité... vont d'abord être contrôlées, pour s'assurer de la similarité avec le produit de référence.
Ensuite, des études pré-cliniques vont être réalisées.
Puis des essais cliniques, de phase I et de phase III, sont menés. L'équivalence de sécurité et d'efficacité sont alors vérifiées.
L'avantage dans le développement d'un biosimilaire, comparé à celui d'un médicament de référence, est que l'objectif consiste à démontrer une équivalence, et non une supériorité.
Aussi, il n'est pas nécessaire de réaliser des essais de phase II (détermination de la posologie à utiliser).
Texte légal : Procédure d'enregistrement
Jusqu'à présent, les médicaments biosimilaires qui ont été développés sont des protéines recombinantes. À ce titre, en Europe, conformément au règlement CE n°726/2004 du Parlement européen et du Conseil du 31/03/2004, les laboratoires désirant mettre sur le marché un biosimilaire doivent suivre la procédure centralisée. Un dossier unique sera analysé par le Comité des médicaments à usage humain (CHMP) de l'Agence Européenne des médicaments (EMA). Les données sont évaluées par les toutes les Agences Nationales d'Evaluation (l'ANSM pour la France).
Cela permettra l'octroi d'une AMM commune à tous les États membres de la Communauté Européenne. Le produit sera accessible à l'ensemble du marché communautaire.
Puis, à l'échelle nationale, le médicament biosimilaire autorisé devra passer par différentes étapes : fixation du prix, demande de remboursement, inscription sur la liste en sus...
Remarque : Vers une simplification du développement des biosimilaires ?
La nécessité de réaliser des essais cliniques permettant d’évaluer l’efficacité et la sécurité est interrogée.
Il est suggéré qu’une comparabilité structurelle et fonctionnelle ainsi que des données pharmacocinétiques pourraient être suffisantes pour prouver la similarité avec le biomédicament de référence.
Un document de réflexion a été mis en ligne ; les parties prenantes ont jusqu’au 30 septembre 2025 pour le commenter.